共价有机框架(COFs),Advanced Materials!
康奈尔大学
Advanced Materials
共价有机框架
VCOFs
2023-12-30 09:22:24
0
2968
光催化已成为可再生能源发电、环境修复和有机合成等应用的强大工具。开发新型高效光催化剂对推进光化学领域的发展至关重要,特别是使化学合成更加高效和可持续。近年来,在开发高效光催化剂方面取得了重大进展。一种方法是使用半导体材料,因为它们可以吸收光能并产生引发化学反应的载流子。其他方法包括设计特定结构和成分的材料,以增强其光催化性能。开发高效光催化剂的主要挑战之一,是寻找能够吸收宽光谱光并产生寿命长的高能载流子的材料。共价有机框架(COFs),具有高孔隙率和极其有序的形态,作为一个多功能平台(用于开发广泛应用的新材料)受到了广泛的关注。COFs由于可调谐性、多孔性和半导体电子特性,已成为有前景的光催化材料。特别是,含有活性氮或碱性组成的COFs,由于其能够响应pH而发生结构变化,已显示出作为酸传感器的巨大潜力。COFs可用于检测大气中酸性气体的存在,如HCl、NO或NO2。与传统传感材料相比,COFs酸传感器具有重量轻、可回收、更高的灵敏度、选择性和耐用性等优点。因此,一种高灵敏度和选择性的COF酸传感器,在准确检测和监测酸水平方面具有巨大潜力。通过Knoevenagel缩合反应的乙烯基连接的COFs(VCOFs),由于其结构中存在共轭亚乙烯键,具有独特的电子和光学性质。因此。VCOFs在催化、光电子、燃料电池和能量转换方面具有潜在的应用价值。VCOFs的一个主要优势,是合成过程简单及设计灵活。总体而言,尽管所有类型COFs都具有独特的性能和潜在的应用,但VCOFs的高稳定性、可调孔径以及独特的电子和光学性能,使其在广泛应用中更具吸引力。然而, VCOFs的各种结构、特点和应用仅被部分开发,对VCOFs研究的报道相对较少。近日,美国康奈尔大学Alireza Abbaspourrad报道了首次合成两种乙烯基四嗪连接的共价有机骨架(COF),TA-COF-1和TA-COF-2,表现出1323和1114m2·g-1的高结晶度和高比表面积,良好的能带位置和适用于光驱动应用的窄带隙。这些优点使TA-COFs能够在芳基硼酸氧化和苄胺的光诱导偶联中,作为可重复使用的无金属光催化剂。此外,TA-COFs显示出酸感应能力,在暴露于HCl溶液、HCl蒸汽和NH3蒸汽时表现出可见和可逆的颜色变化。TA-COFs的性能优于先前报道的各种COF光电阴极。COF骨架中的四嗪连接体代表了COF合成领域的重大进步,增强了电荷载流子在光反应过程中的分离效率,并有助于其光电阴极性质。TA-COFs还可以在光催化过程中20min内降解5-硝基-1,2,4-三唑-3-酮(NTO),一种存在于工业废水中的不敏感炸药。因此,揭示了质子化的TA-COFs作为光降解和Brønsted酸催化剂的双重功能。这项开创性工作为四嗪连接体在COF材料中的潜力开辟了新的途径,促进了催化、传感和其他相关领域的进展。相关研究工作以“Tetrazine-Linked Covalent Organic Frameworks with Acid Sensing and Photocatalytic Activity”为题发表在国际顶级期刊《Advanced Materials》上。使用具有C3对称性的两种单体醛(TFPB)(TA-COF-1)和(TFBPB)(TA-COF-2)以及具有D2h对称性的DMTAZ,通过Knoevenagel缩合反应合成了TA-COFs(方案1)。醛形成了整个六边形结构的角,四嗪形成了边缘连接。最佳反应条件为均三甲苯/1,4-二恶烷/乙腈/三氟乙酸的混合溶剂体系在120℃反应7d,可产生最高结晶度。
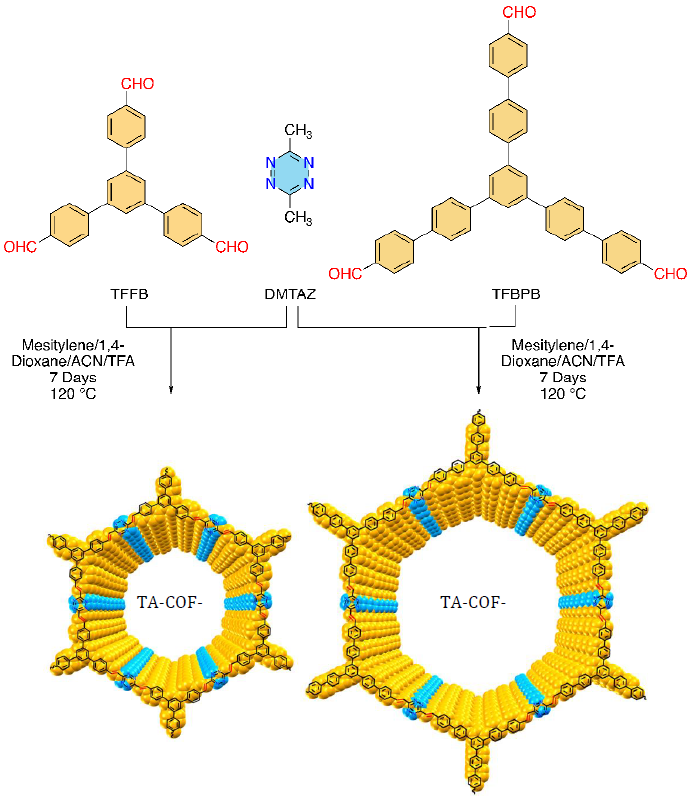
方案1. Knoevenagel缩合合成TA-COF-1和TA-COF-2反应
使用粉末X射线衍射(PXRD)评估了两种TA-COFs的结晶度,尖锐峰的出现证实了它们的高结晶度。两种TA-COFs都显示出五个主要Bragg衍射峰:对于TA-COF-1,峰出现在2.91°、4.99°、5.69°、7.58°和25.34°;对于TA-COF-2,峰值出现在1.98°、3.42°、4.18°、5.27°和24.9°(图1a、b)。这五个峰对应(100)、(110)、(200)、(210)和(002)平面。其中,TA-COF-1和TA-COF-2的最强峰分别位于2θ=2.9°和1.98°,对应于(100)平面。在2θ=25.3°(TA-COF-1)和24.9°(TA-COF-2)处的峰被归属于(002)平面,显示层之间的π-π堆叠。通过遵循布拉格定律的d间距测量,预估TA-COF-1和TA-COF-2相邻片之间的层间距离分别为约0.37和0.38nm。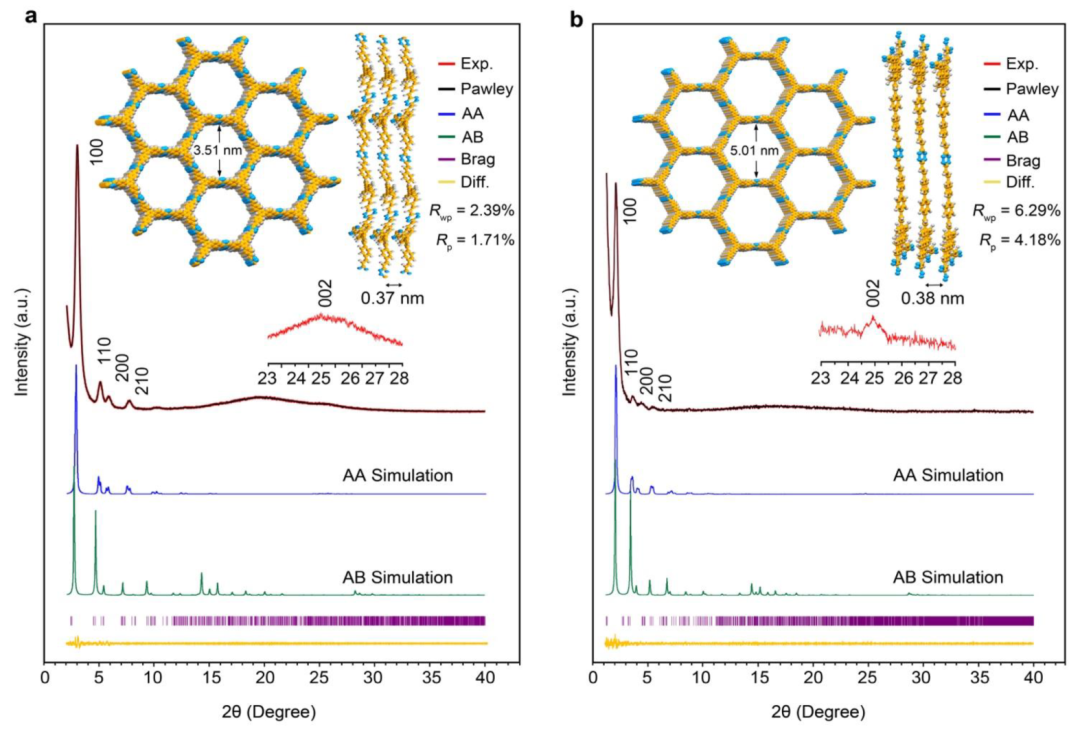
图1. TA-COF-1(a)和TA-COF-2(b)的X射线衍射图13C CP-MAS NMR光谱证实了烯烃和芳族碳的存在,以及醛基的消失(图2)。在TA-COFs的光谱中,四嗪环碳的相关峰出现在167.2ppm。在115-146ppm范围内的宽峰,对应于它们的芳基和烯烃基团。BPVT是由DMTAZ和BPCA在醇/KOH中,通过缩合反应合成(方案2)。TA-COFs的固态13C NMR光谱与模型化合物BPVT的液体13C NMR存在良好的一致性。
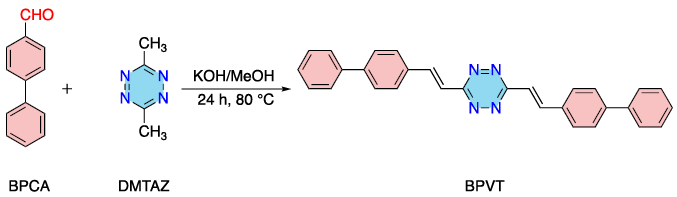
方案2. BPVT的合成
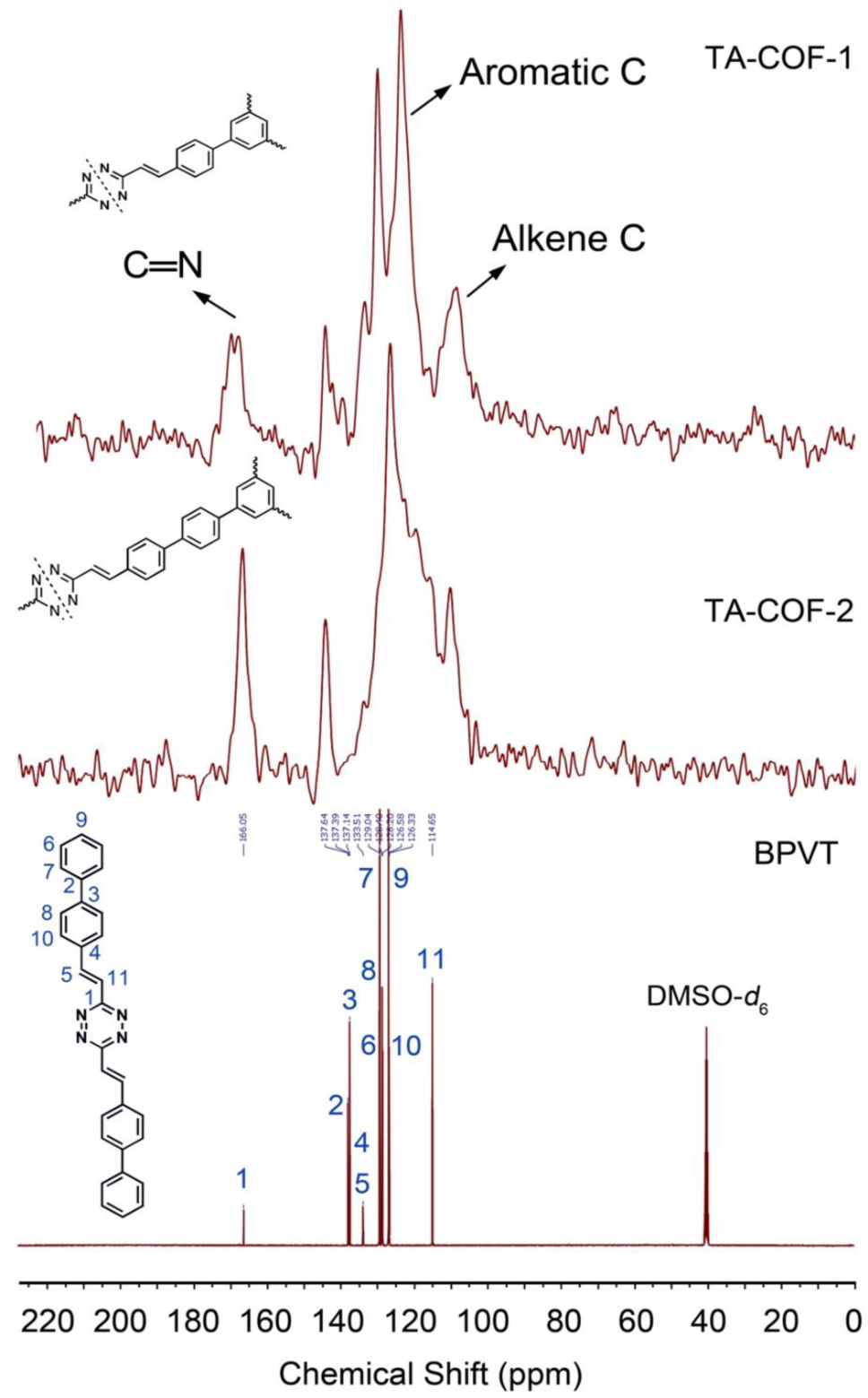
图2. TA-COF-1和TA-COF-2的13C CP-MAS NMR光谱
图3显示,TA-COFs中存在微孔,孔径在纳米级范围内,特别是在1-5nm范围内。该数据为COFs的整体孔隙率和表面积特性提供了至关重要的见解。结果表明,COFs孔隙率的全面图像,包括中孔和微孔区域。在FE-SEM图像中观察到两种TA-COFs的高度多孔形态(图4a-d)。结果表明,TA-COF-1自组装成花朵状,而TA-COF-2自组装成簇状。从图4e、f中可观察到,具有六边形蜂窝状通道的排列良好的层状形态。沿着(100)晶轴的六方孔,验证了2D TA-COF结构的AA堆叠模式(图4e)。此外,观察到具有0.40 nm周期的精确组织的帧边界(图4f)。这种周期性对应于(002)平面,与PXRD图案中在2θ=25.3°时d值为0.37nm一致。图4f 中FFT图像显示了以六边形排列的白点图案,表示存在以六边形组织的(100)小平面的孔。从选区电子衍射(SAED)中观察到五个不同的衍射环,(100)、(110)、(200)、(210)和(002),对应于TA-COFs晶体内的晶面(图4g)。
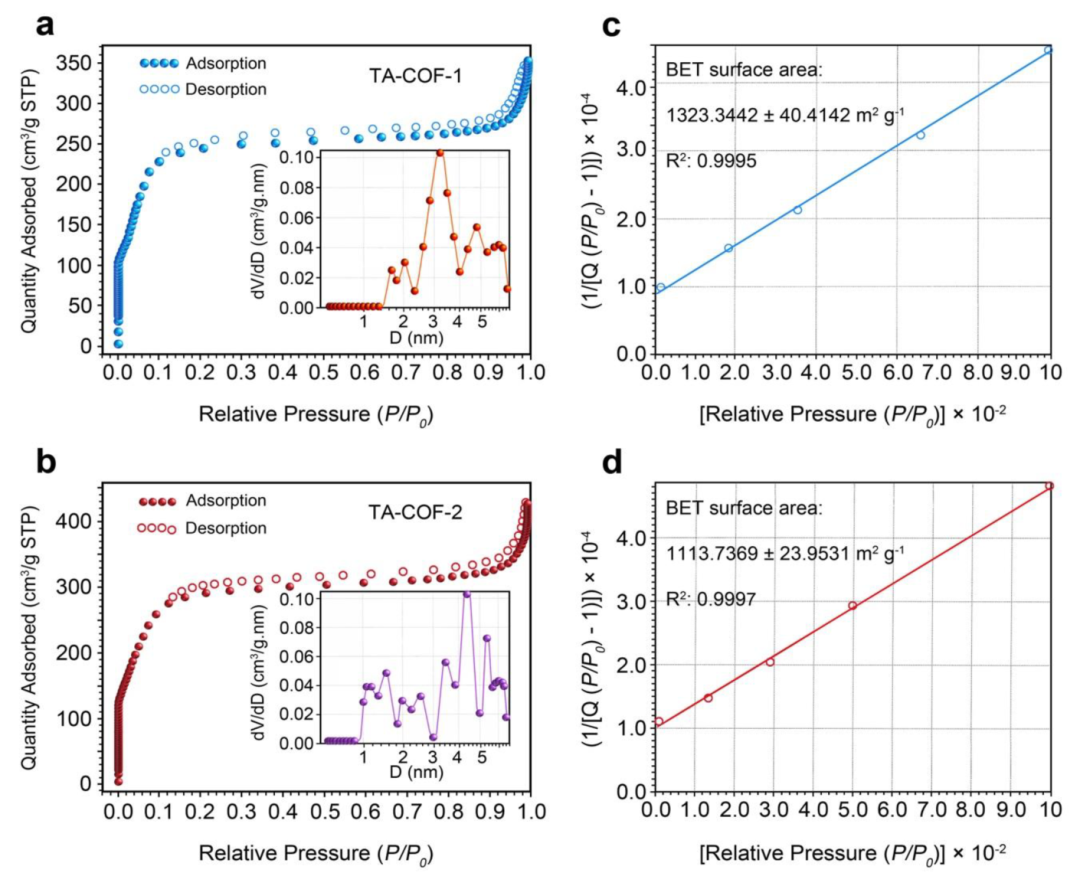
图3. TA-COF-1和TA-COF-2的氮吸附解吸等温线和多点BET拟合
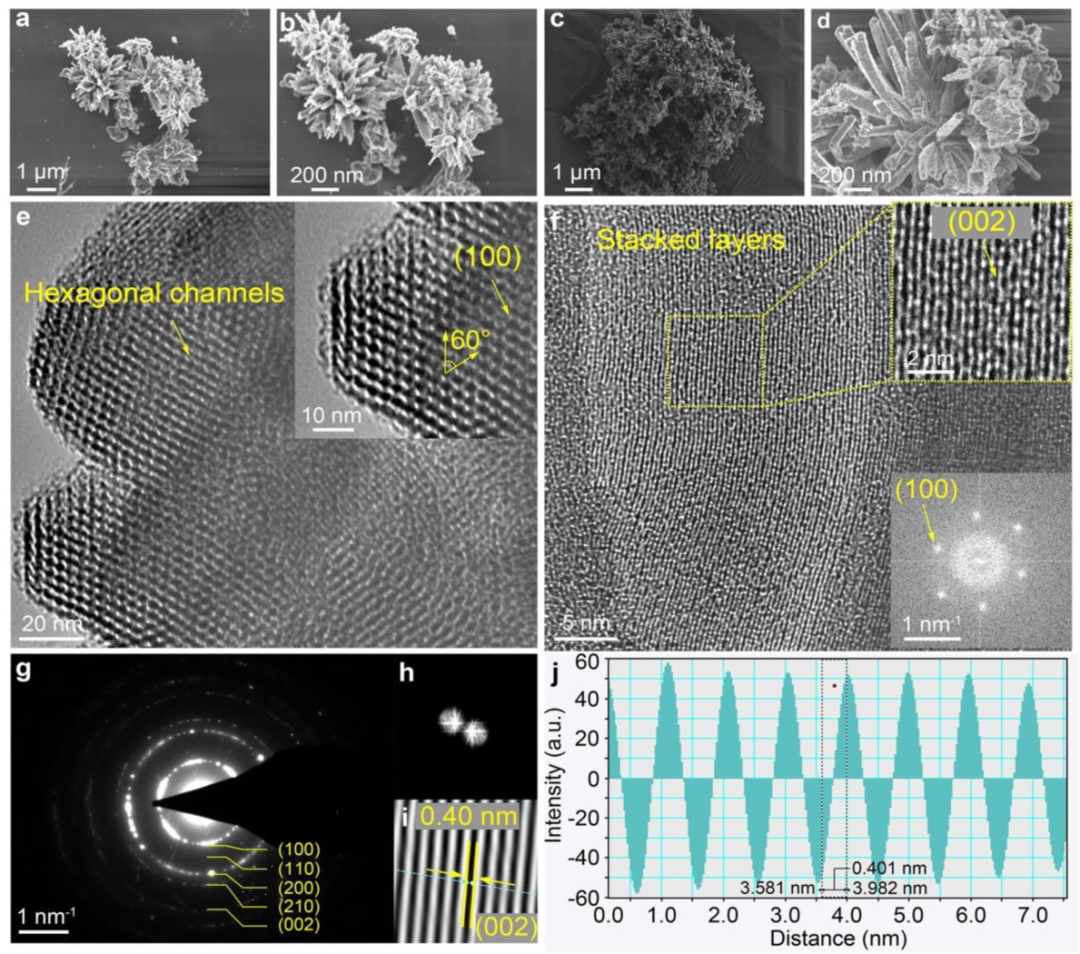
图4. TA-COFs的FE-SEM图像、HRTEM图及SAED图
使用UV-vis漫反射和PL光谱,探测光催化过程中TA-COFs内的电子跃迁。UV-vis中,TA-COF-1和TA-COF-2分别在438nm和412nm处显示最大值。此外,400~600nm的宽峰,表明TA-COFs可以在大范围的可见光区域内捕获光子(图5a)。对TA-COF-2,TA-COF-1(590nm)的吸收边值红移约20nm(570nm)。根据可见光反射率数据和Kubelka-Munk方程,TA-COF-1的光学带隙为(𝐸𝑔) 为2.65eV,TA-COF-2为2.78eV(图5b)。在365nm的照射下,TA-COF-1粉末发黄光,TA-COF-2粉末发绿光。在365nm激发时,TA-COF-1在588nm处表现出最大发射。然而,TA-COF-2在565nm处显示出略短的最大发射,可归因于其苯结的数量较多,导致π-电子在整个框架中的传播更加明显地减少(图5c)。两种荧光衰减都显示出良好拟合的指数衰减行为(图5d)。根据PL衰减曲线,TA-COF-1和TA-COF-2的平均寿命分别为4.31和3.23ns。TA-COF-1的窄带隙有助于延长其荧光寿命。
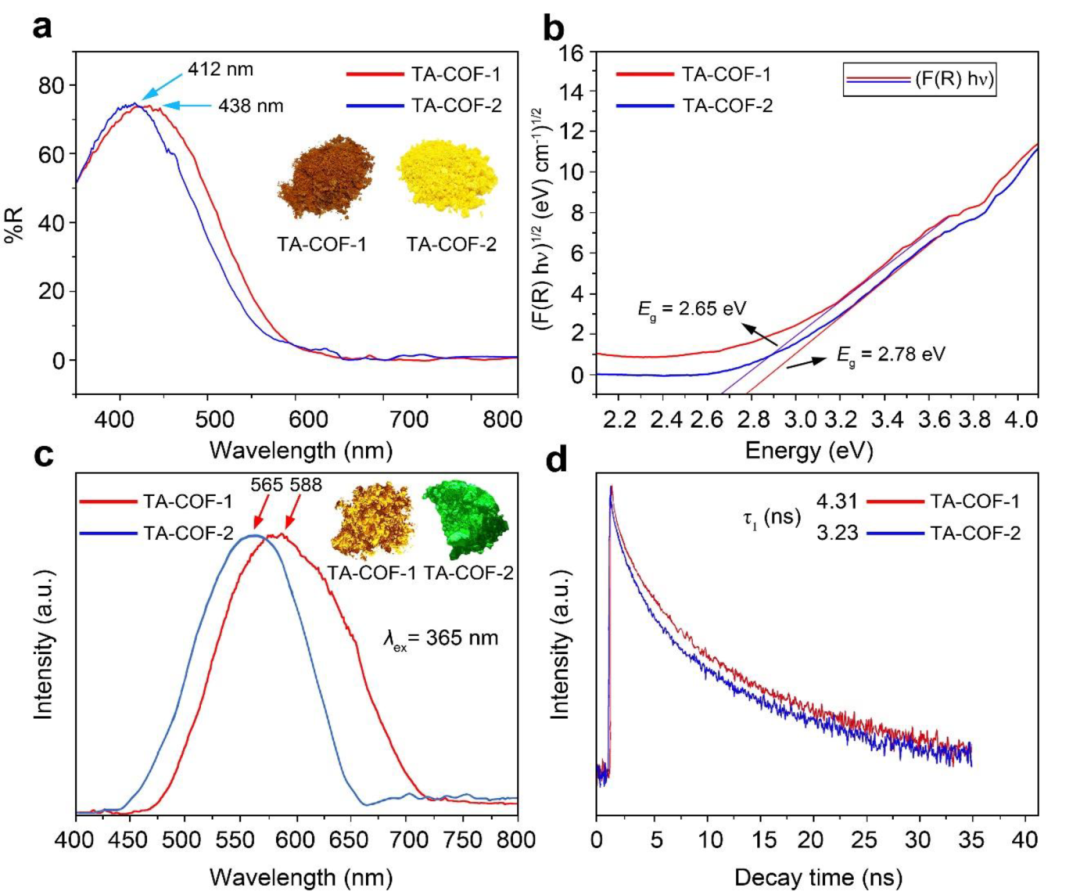
图5. TA-COFs的固态UV-vis漫反射(a)、光学带隙(b)、荧光光谱(c)、荧光衰减指数时间拟合扫描(d)
进一步研究了TA-COFs的酸敏感性。TA-COFs在1,4-二恶烷(0.1mg·mL-1)中的悬浮液,并在暴露于不同浓度HCl中测量荧光。向TA-COF-1悬浮液中加入10μL HCl(5.0μmol·L-1),导致507和525nm处的峰强度减弱,而556nm处出现新峰(图6a)。随着HCl浓度从6.0增加到59.6μmol·L-1,556nm处的峰强度稳步增加,59.6μmol·L-1后没有明显变化。COFs的荧光强度与HCl浓度存在线性关系(R2=0.96862)。使用外推法,发现最低检测限为4.1×10-3μmol·L-1(图6b)。
根据荧光衰减曲线,TA-COF-1在HCl溶液(59.6μmol·L-1)中的寿命为1.33ns(图6c)。HCl传感的一个合理机制表明,在酸性条件下,每个四嗪环上的氮杂原子被质子化。四嗪的质子化导致π-π*跃迁极大值从λmax 295到336nm的显著红移。这是由于碱及其相应共轭酸的电子结构之间的显著差异。电子能级的实质性变化,导致颜色变化(图6d)。TA-COFs对酸性和碱性蒸汽也非常敏感。当暴露于HCl蒸汽时,观察到TA-COF-1从橙色到深棕色,及TA-COF-2从黄色到橙色的可逆颜色变化。暴露于氨蒸气后,COFs恢复到其原始颜色(图6e)。TA-COFs对HCl气体的反应在数周内保持不变。在延长的实验中,尽管重复暴露循环,但没有观察到酸感应能力的损失。
图6. 酸处理TA-COF-1悬浮液的荧光光谱(a)、荧光强度与酸浓度的相关曲线(b)、PL衰减光谱(c)、TA-COFs上可能的质子化和去质子化位点(d)、TA-COFs粉末暴露于HCl和NH3蒸汽后的裸眼照片(e)采用电化学方法研究了TA-COFs的各种电子特性。FE-SEM图像表明了薄膜的均匀多孔纹理,其厚度为几微米(图7a)。TA-COF-1和TA-COF-2在0.2M Na2SO4水溶液(pH=6.8)中的CV曲线,在多次扫描过程中显示出可逆的氧化和还原过程,表明具有良好的电化学稳定性(图7b)。斜率的正曲线表示n型半导体的典型行为,并且存在于两种TA-COFs中,其中电子是主要载流子(图7c)。与正常氢电极(NHE)相比,TA-COF-1和TA-COF-2在pH=6.8时𝐸𝑓𝑏分别为-1.45 V和-1.61 V。通过测量TA-COF-1(图7d)和TA-COF-2的VBM水平,发现与真空水平相比,VBM水平分别为-6.00和-6.27eV。TA-COF-1和TA-COF-2的能带图表明,三种成分的能级是一致的,使得对于将O2还原为超氧化物自由基,CBM比O2的Ered负得多(图7e)。TA-COFs光电极的斩波电流-电势曲线,在进行光开关时显示出令人印象深刻的光响应(图7f)。相对于可逆氢电极(RHE),当施加0.8 V的偏置电位时,检测到阴极光电流,TA-COF-1的饱和电流密度约为27μA·cm-2,高于TA-COF-2(15μA·cm-2)。TA-COF-1表现出最高的光电流响应,表明内部电荷转移更快。
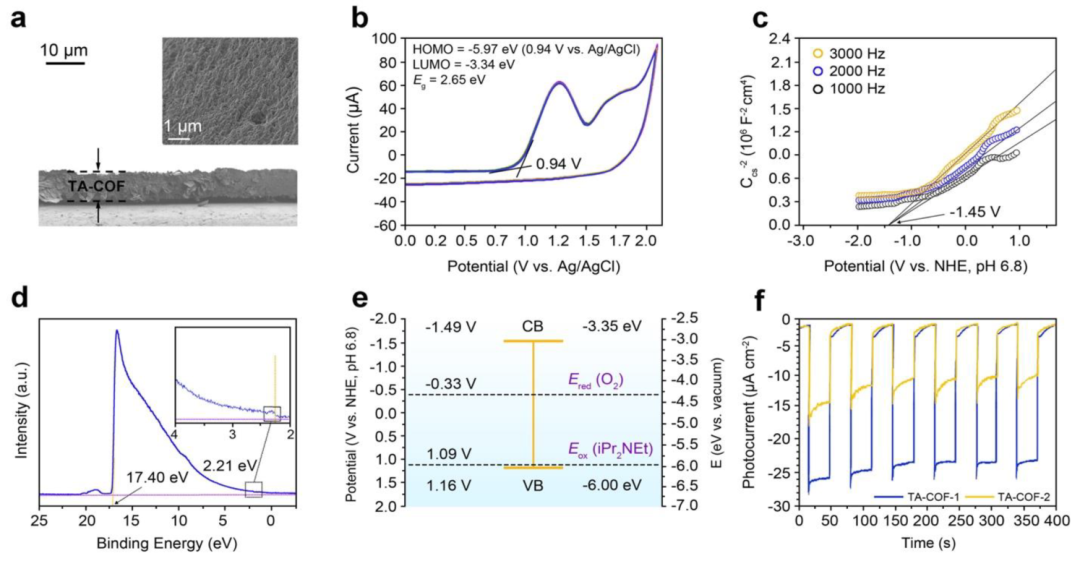
图7. 滴铸TA-COF-1膜的横截面和顶视图(插图)FE-SEM图像(a)、TA-COF-1与Ag/AgCl(+0.199 V)的CV曲线(b)、TA-COF-1膜的Mott-Schottky图(c)、TA-COF-1的UPS图谱(d)、TA-COF-1的能量图(e)、斩波阴极光电流密度与时间的关系(f)
通过研究NTO降解时间,评估了质子化和未质子化TA-COFs对阳光驱动的光解效率的影响(图8)。在没有任何光催化剂的情况下,NTO在阳光照射下不会分解。与未质子化相比,TA-COF-1-H+和TA-COF-2-H+对NTO的光降解能力增强。在TA-COF-1-H+中,暴露于阳光下20min,观察到NTO的浓度迅速下降;而TA-COF-2-H+中,则需要25min(图8a)。与质子化TA-COFs相比,未质子化TA-COFs显示出略低的光催化活性(图8b)。图8c显示,与未质子化TA-COFs相比,质子化TA-COFs表现出明显的光催化能力增加。TA-COF-1-H+的光降解效率最高,表观速率常数为0.21min-1,是TA-COF-1(0.05min-1)的四倍,是TA-COF-2(0.04min-1)的五倍多。
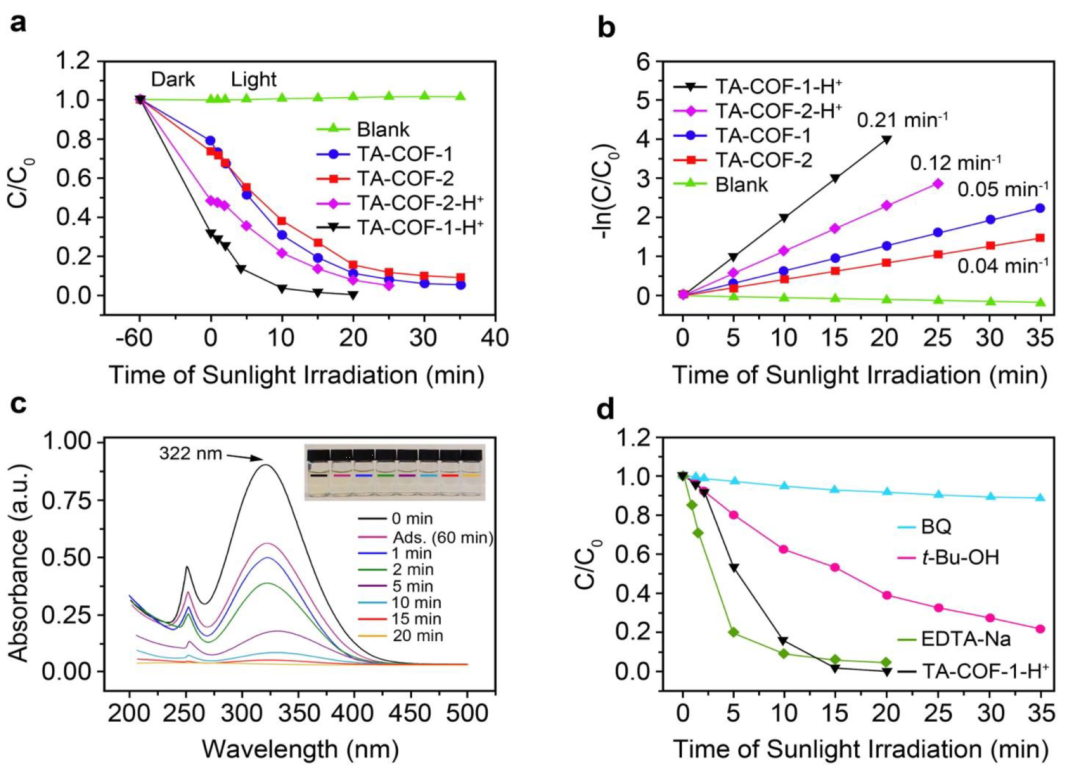
图8. 使用质子化和未质子化的TA-COFs光催化剂,阳光驱动的NTO光催化降解(150mg·L-1)
总之,这项研究通过使用公认的合成策略,成功制备了两种乙烯基四嗪连接的COFs,其具有优异的结晶度、高表面积和明确的π-共轭框架。这些COFs显示出高的光活性和相当小的HOMO-LUMO间隙,还表现出显著的化学稳定性,对强酸(HCl 6M)、腐蚀性碱(NaOH 6M)和长时间暴露在光下7天表现出优异抗性。高光活性和化学稳定性的结合,使这些COFs成为广泛用于需要有效催化作用和对恶劣环境有抵抗力应用的有前景的候选者。作为光催化剂,TA-COFs为光辅助氧化反应提供了一种无金属的选择,如芳基硼酸羟基化为相应的酚,以及取代苄胺氧化偶联为N-亚苄基苄基胺。在光催化过程中,反应转化率均>90%,且通过PXRD监测,COFs保持结晶度。此外,它们很容易回收,且在12次催化循环后,结晶度仅发生微小变化。所得COFs在评估材料中表现出最高的光电流响应和光电导性。这些结果强调了TA-COFs作为各种应用中高效光电阴极的显著潜力。TA-COFs对盐酸敏感,可通过响应不同HCl浓度的荧光强度和发射波长的显著变化来证明。TA-COFs对不同酸蒸汽的反应,突出了它们的选择性,因为它们仅在暴露于HCl蒸汽时发生可逆的颜色变化,从而将其与其他酸区分开。TA-COFs在酸性和碱性条件下都表现出可逆的颜色变化,使其成为有效的HCl酸传感器。它们的快速“开启”荧光反应,肉眼可见,发生不到一秒钟。还表明了,通过利用质子化的TA-COFs的双重性质(作为光催化剂和Brønsted酸),TA-COFs可以在光照下快速有效降解NTO。质子化TA-COFs显著加快了光解速率,导致NTO在20min内完全分解。据我们了解,这是COF在NTO催化光解中的首次应用。这项研究成果有助于网状设计原理的不断发展,从而能够创建为太阳能收集和转换量身定制的高效和高活性的COF系统。COF结构中四嗪单元的固有特性促进了这一进展。TA-COFs材料的多功能性和性能,为扩大COFs在不同领域的应用范围和应对未来的各种技术挑战开辟了机会。文献链接:
https://doi.org/10.1002/adma.202311042
本文为e测试原创,未经允许,禁止转载!






















